


醫療器械創新網

12月1日,國家藥監局發布《抗腫瘤藥物的非原研伴隨診斷試劑臨床試驗注冊審查指導原則》《使用體外診斷試劑境外臨床試驗數據的注冊審查指導原則》以及《神經和心血管手術器械-刀、剪及針注冊審查指導原則》。
01/抗腫瘤藥物的非原研伴隨診斷試劑臨床試驗注冊審查指導原則

伴隨診斷試劑臨床試驗目的主要包含兩個方面,一方面為確認試劑臨床性能,另一方面為確認伴隨診斷用途。根據伴隨診斷試劑設計開發的特點,確認其伴隨診斷用途的臨床研究可分為如下幾種情況:
(一)如為原研伴隨診斷試劑,可提交該產品作為伴隨診斷試劑參與的藥物臨床試驗資料作為確認伴隨診斷用途的臨床試驗資料,或提交與藥物臨床試驗中所使用的CTA進行橋接試驗的臨床試驗資料。具體可參考與抗腫瘤藥物同步研發的原研伴隨診斷試劑臨床試驗的相關要求。
(二)申報產品如為非原研伴隨診斷試劑,其伴隨診斷用途可根據具體情形采用下列適用的方式之一進行研究:與原研伴隨診斷試劑進行一致性比對、橋接試驗、已上市抗腫瘤藥物療效的觀察性研究。
針對所伴隨的抗腫瘤藥物已上市多年、臨床應用廣泛、意義明確、判讀易于標準化的伴隨診斷試劑,如申報產品的性能與原研伴隨診斷試劑具有較好的可比性,則申報產品伴隨診斷用途的確認可采取與原研伴隨診斷試劑進行一致性比對的方式,此類生物標志物清單見表1。申請人擬開發的未包含在表1中的生物標志物,如有需要可與監管部門充分溝通后確定其伴隨診斷臨床意義。
申報產品所檢測的生物標志物中存在針對抗腫瘤藥物療效負性選擇的生物標志物。例如RAS基因,已批準的西妥昔單抗說明書中明確載明,該藥物不用于RAS基因的突變的結直腸癌患者。針對此類生物標志物,申報產品伴隨診斷用途的確認可采取與原研伴隨診斷試劑或CTA進行一致性比對的方式,臨床試驗應重點關注二者的一致性。
表1.相關生物標志物清單

注:基于當前認知及我國相關產品的開發及臨床應用情況,相關抗腫瘤藥物已上市、臨床應用廣泛、意義明確、判讀易于標準化并且臨床應用多年的伴隨診斷生物標志物清單見表1。該清單會隨著科學認知的深入及相關產品的臨床應用情況適時更新。
02/使用體外診斷試劑境外臨床試驗數據的注冊審查指導原則
本指導原則旨在為申請人使用體外診斷試劑境外臨床試驗數據在我國進行注冊申報提供指導,適用于進行首次注冊申報和相關變更注冊申請的產品。
本指導原則聲稱的境外臨床試驗數據是指,全部或同期在境外具備臨床試驗開展所在國家(地區)要求條件的臨床試驗機構中,對擬在我國注冊申報的體外診斷試劑進行臨床試驗時所產生的研究數據。
差異分析導圖
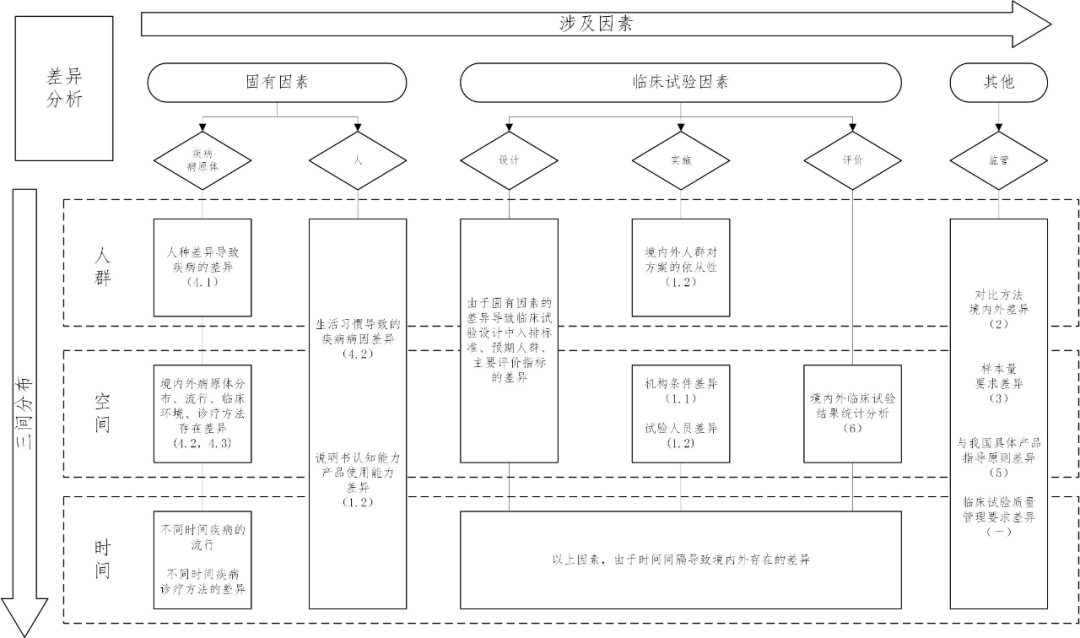
注:1.圖中每部分內容均不是對該部分內容的窮舉,應當根據產品的特性對境內外的差異進行具體分析。
2.不同因素、三間分布之間可能存在交叉。
3.表格主要以“(二)境內外臨床試驗設計關鍵要素的差異分析”章節為基礎。
03/神經和心血管手術器械-刀、剪及針注冊審查指導原則
本指導原則適用于第二類神經和心血管無源手術器械中的手術刀、手術剪及手術針。
產品技術要求
產品技術要求的制定應符合《醫療器械產品技術要求編寫指南》的要求。注冊申請人需根據產品的技術特征和臨床使用情況來確定產品的性能指標和檢驗方法。對注冊申請人宣稱的產品的所有技術參數和功能,若適宜,均需在產品技術要求中予以規定。產品技術要求中的試驗方法均需為已經過驗證的方法。若對標準中的試驗方法有所修改,需明確修改的內容和原因,并在研究資料中提供驗證資料。對于相關行業標注、國家標準中不適用的要求條款,需說明不適用的原因和依據。
(1)產品型號/規格及其劃分的說明
列表說明產品的型號、規格,明確產品的型號、規格的劃分說明,鑒于該類器械型號、規格較多,建議在附錄中以列表的形式提供,列表中需明確具體組件的種類和數量。
(2)性能指標
產品性能研究項目中,對于可進行客觀判定的成品的功能性、安全性指標,需將其列入產品技術要求。可包括但不限于以下性能:外觀、尺寸、硬度、表面粗糙度、耐腐蝕性能、連接牢固度(適用于有連接部件的產品)、使用性能(可客觀判斷的性能,如刀片刃口鋒利度、手術剪的剪切性能等)、環氧乙烷殘留量(適用于環氧乙烷滅菌的產品)、無菌(適用于滅菌狀態交付的產品)。
(3)檢驗方法
產品的檢驗方法需根據性能指標設定,檢驗方法需優先采用公認的或已頒布的標準檢驗方法;自建檢驗方法需提供相應的方法學依據和理論基礎,同時保證檢驗方法具有可操作性和可重現性,必要時可附相應圖示進行說明,文本較大的可以附錄形式提供。
(4)附錄
建議注冊申請人以資料性附錄形式提供產品的結構圖示及制造材料信息。

